



ABOVE PHOTO: (Photo/Shutterstock)
By Matthew Perrone
ASSOCIATED PRESS
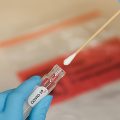
WASHINGTON — U.S. regulators on Tuesday allowed emergency use of the first rapid coronavirus test that can be performed entirely at home and delivers results in 30 minutes.
The announcement by the Food and Drug Administration represents an important step in U.S. efforts to expand testing options for COVID-19 beyond health care facilities and testing sites. However, the test will require a prescription, likely limiting its initial use.
The FDA granted emergency authorization to the single-use test kit from Lucira Health, a California manufacturer.
The company’s test allows users to swab themselves to collect a nasal sample. The sample is then swirled in a vial of laboratory solution that plugs into a portable device. Results are displayed as lights labeled positive or negative.
To date, the FDA has authorized nearly 300 tests for coronavirus. The vast majority require a nasal swab performed by a health professional and must be processed at laboratories using high-tech equipment. A handful of tests allow people to collect their own sample at home — a nasal swab or saliva — that’s then shipped to a lab, which usually means waiting days for results.
Health experts have called for options to allow people to test themselves at home, reducing turnaround times and the potential spread of the virus to others, including health care workers. Rapid test results are critical to quickly quarantining those who are infected and tracing their contacts. But for months, U.S. testing has been plagued by slow results due to bottlenecks as testing laboratories. There are other rapid tests but most require a small, special machine operated by a health professional to develop results
“Now, more Americans who may have COVID-19 will be able to take immediate action, based on their results, to protect themselves and those around them,” Dr. Jeff Shuren, director of the FDA’s devices center, said in a statement.
Lucira did not immediately respond to a request for additional details after business hours Tuesday.
The Lucira COVID-19 test grew out of research the company was doing to develop an at-home flu test, according to the company’s website. Lucira adapted its technology to detect COVID-19 after the outbreak.
The test uses technology similar to genetic laboratory-based tests that are the standard tool for COVID-19 screening. That’s different than most rapid tests currently used in the U.S., which look for viral proteins called antigens — not the virus itself.
Anyone that tests positive should isolate and seek care from a health professional, the FDA said in its release. Those who test negative but still have coronavirus symptoms should consult a doctor; a negative result does not rule out COVID-19 infection.
The FDA said Lucira’s test was also authorized for use in doctor’s offices and testing sites. Currently all U.S. testing sites must report results to state and federal health authorities tracking the pandemic. Doctors will be required to report the home test results.
“If the results are not reported back, it may be difficult to figure out what is happening in the community at large,” Dr. Alberto Gutierrez, former head of the FDA’s testing office, said in an interview before the announcement.
More than two dozen companies have been racing for months to develop the first, rapid home-based test for COVID-19. However, the FDA outlined a number of study requirements for manufacturers.
These hurdles have less to do with COVID-19 specifically, and more to do with decades-long concerns about whether people without any medical training can accurately screen themselves and interpret the results.
The FDA has only ever approved one home test for an infectious disease — an HIV test. And even commonplace over-the-counter tests— such as home pregnancy kits — were subject to years of scrutiny before FDA allowed their use in the 1970s.
Experts say that careful approach is warranted for coronavirus.
“I think increased testing closer to patients, including in the home, is the way of the future,” Dr. Robin Patel of the Mayo Clinic said in an interview before the announcement was made. “But there are considerations that have to be addressed to make sure that this is done in a safe and effective way.”
FDA regulators authorized the new test using their emergency powers to quickly speed the availability of experimental products during public health crises. In normal times, the FDA requires evidence of safety and effectiveness before clearing a new test. But during public health emergencies the agency can lower those standards.
The FDA release did not disclose the test’s accuracy or the study results that regulators used to make the decision.









Leave a Comment